Jednym z największych wyzwań w leczeniu tocznia jest niejednorodny (heterogenny) obraz kliniczny pacjentów.1 Mówi się obrazowo, że pacjenci z toczniem są „jak płatki śniegu” – nie ma dwóch takich samych. Poniższy artykuł podsumowuje przyczyny różnorodności tej choroby z uwzględnieniem czynników chorobotwórczych i patomechanizmów immunologicznych.
Kliniczna i immunologiczna heterogenność SLE (ang. Systemic Lupus Erythematosus)
Na obraz kliniczny chorego na toczeń składa się kombinacja indywidualnych o objawów klinicznych, stopnia ciężkości choroby i kierunku jej progresji. Różnorodność manifestacji klinicznych jest wynikiem oddziaływania uwarunkowań genetycznych danej osoby z czynnikami środowiskowymi, w wyniku którego dochodzi do powstania całego spektrum zaburzeń immunologicznych, takich jak dysfunkcja komórek układu odpornościowego (limfocytów T i B, neutrofilów, komórek mieloidalnych), zmiany w profilach wydzielanych cytokin lub nasilenie odpowiedzi zależnej od interferonów typu I.1
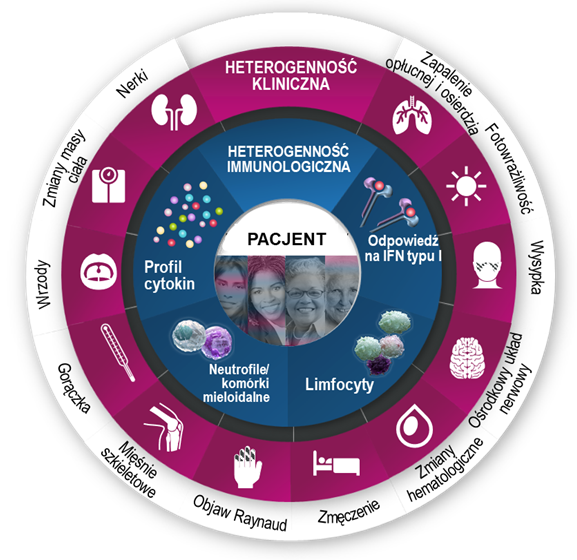
Obraz pacjenta z toczniem jest wypadkową jego uwarunkowań genetycznych, czynników chorobotwórczych i aktywowanych patomechanizmów immunologicznych.
Cechy chorobotwórcze wpływające na heterogenność populacji pacjentów z toczniem.
Badania nad patofizjologią tocznia skupiały się do tej pory głównie na limfocytach B, ponieważ komórki te są źródłem autoreaktywnych przeciwciał z klasy IgG, a to właśnie obecność tych przeciwciał jest cechą charakterystyczną tocznia. Natomiast rola innych elementów wrodzonej i nabytej odpowiedzi odpornościowej w patomechanizmach tocznia została opisana stosunkowo niedawno. Należą do nich: 2,3
- czynniki środowiskowe (w tym światło UV), wirusy, inne patogeny, elementy strukturalne mikroorganizmów, niektóre leki, czynniki hormonalne i te związane z chromosomem X
- czynniki genetyczne i epigenetyczne: lista wariantów genowych i modyfikacji epigenetycznych (np. hipometylacja DNA, microRNA) związanych ze zwiększonym ryzykiem wystąpienia SLE cały czas się powiększa
- nasilone procesy NETozy i upośledzone usuwanie produktów rozpadu komórek, w tym kompleksów immunologicznych zawierających kwasy nukleinowe
- zaburzone mechanizmy wrodzonej odpowiedzi odpornościowej oraz nasilona produkcja interferonów typu I przez komórki NK, neutrofile, makrofagi czy komórki dendrytyczne
- zaburzone mechanizmy odpowiedzi nabytej: nieprawidłowa prezentacja i rozpoznawanie autoantygenów oraz związana z tym nieprawidłowa odpowiedź ze strony limfocytów T i B
- przewlekły stan zapalny napędzany przez cytokiny prozapalne, interferony i produkty procesu NETozy
Patofizjologię tocznia podsumowuje wideo, które można znaleźć tutaj.
NEToza to programowana śmierć komórki, w wyniku której neutrofile uwalniają zewnątrzkomórkowe pułapki (ang. neutrophil extracellular traps, NETs) składające się z chromatyny i białek o właściwościach antybakteryjnych. Struktury uwalniane w procesie NETozy fizycznie unieruchamiają mikroorganizmy i potencjalnie prowadzą do ich zabicia. Jednak elementy tych struktur mogą aktywować inne komórki układu odpornościowego.2
Progresja mechanizmów chorobotwórczych w toczniu.
Na patogenezę tocznia i progresję choroby do momentu rozpoznania wpływ ma wiele aspektów. Czynniki genetyczne i środowiskowe przyczyniają się do zaburzenia autotolerancji, w rezultacie czego dochodzi do zaburzenia odpowiedzi immunologicznej.4 Ta, z początku prawidłowa, z czasem może zostać skierowana przeciwko własnym antygenom, co skutkuje tworzeniem autoreaktywnych przeciwciał, cytokin i interferonów. Procesy te mają z początku charakter łagodny i nie dają objawów, dlatego też sama obecność wymienionych markerów znacznie poprzedza diagnozę tocznia.5 Wykazano, że obecność przeciwciał przeciwjądrowych ANA i podwyższony poziom aktywności interferonów typu I obserwuje się nawet kilka lat przed pojawieniem się objawów klinicznych.6,7 U niektórych osób, prawdopodobnie w odpowiedzi na wtórny uraz prozapalny, dochodzi do nasilenia procesów autoagresji i zwiększenia produkcji autoreaktywnych przeciwciał, cytokin i interferonów. Czynniki te z kolei aktywują komórki układu odpornościowego, dochodzi do akumulacji uszkodzeń tkanek i w efekcie - pełnej manifestacji klinicznej choroby.4,7
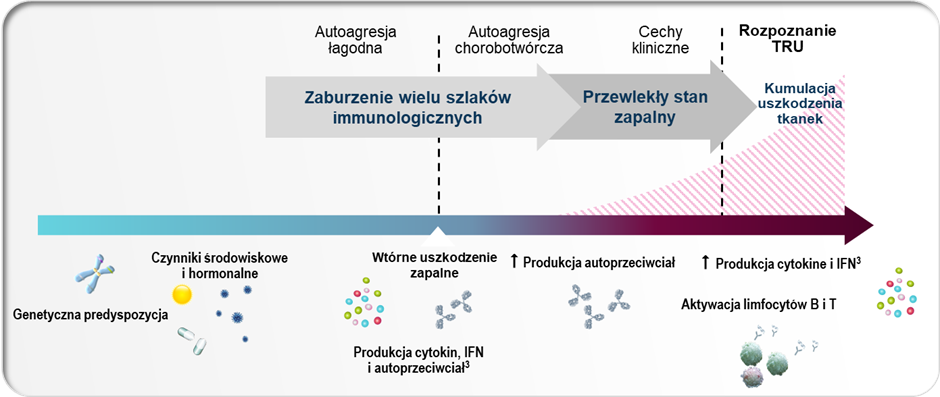
Wielowymiarowa patogeneza i progresja choroby do momentu postawienia rozpoznania.
Interferony typu I a obraz kliniczny pacjentów z SLE.
Pierwsze doniesienia o roli interferonów typu I w patogenezie SLE pochodzą z lat 80tych ubiegłego wieku – zaobserwowano wtedy podwyższony poziom tych cytokin w osoczu pacjentów z aktywną postacią choroby.8,9 Badania in vitro przeprowadzone na początku XXI wieku wykazały, że IFNα obecny w osoczu pacjentów indukuje różnicowanie monocytów w kierunku komórek dendrytycznych, które są zdolne do prezentacji autoantygenów limfocytom T.10 Rozwój technologii umożliwiającej analizę całego genomu umożliwił w 2003 r. różnym grupom badawczym wykazanie, że we krwi obwodowej pacjentów z SLE ekspresja genów należących do sygnatury genowej IFN typu I jest zwiększona.11,12,13,14 Taką cechę stwierdza się u 60% do 80% wszystkich dorosłych pacjentów z umiarkowaną do ciężkiej postacią choroby.15,16,17,18 Więcej o sygnaturze genowej IFN typu I można przeczytać w dalszej części artykułu. Ponadto podanie rekombinowanego IFNα pacjentom chorym na nowotwory lub wirusowe zapalenie wątroby typu C może prowadzić do powstania przeciwciał przeciwjądrowych i rozwoju objawów choroby podobnej do tocznia.19 Co ciekawe, podwyższony poziom aktywności interferonów typu I obserwuje się nawet kilka lat przed pojawieniem się klinicznych objawów choroby.6,7
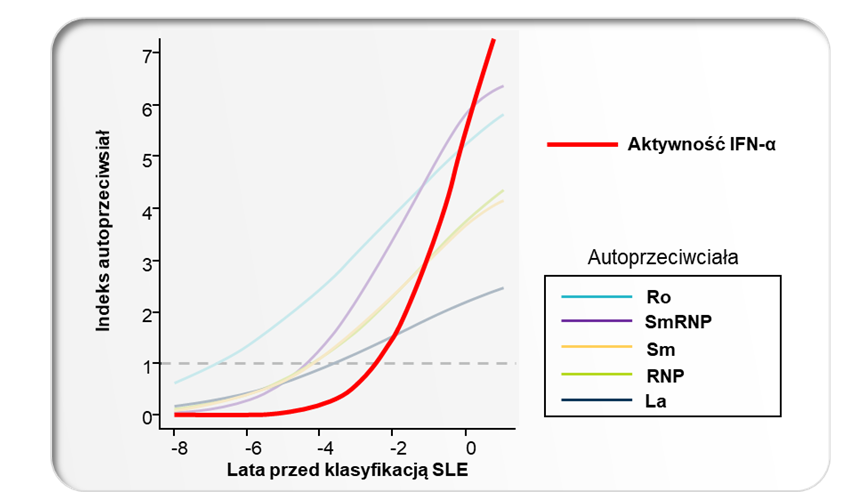
Zmiany poziomu autoprzeciwciał i aktywności cytokin w osoczu poprzedzające diagnozę SLE (n=55);
IFN = interferon; La = antygen toczniowy; RNP = rybonukleoproteina; Sm = anti-Smith.
Aktywność IFN typu I w bezpośredni sposób przekłada się na obraz kliniczny pacjentów z SLE, niezależnie od zajętego narządu:
- skóra: przewlekły charakter manifestacji skórnej tocznia jest zależy od cyklicznej reaktywacji szlaków wrodzonej odpowiedzi odpornościowej. Odpowiedzialne są za to mechanizmy należące do odpowiedzi nabytej.20 W cyklu tym kluczową rolę pełnią keratynocyty, które wydzielają interferony typu I i typu III (takie jak IFNκ czy IFNλ) oraz zależne od interferonów cytokiny i chemokiny. Ponadto w obrębie zmian skórnych u pacjentów z SLE obserwuje się zwiększoną aktywność różnych izoform interferonów typu I, takich jak IFNα, IFNβ czy IFNκ.21,22
- stawy: w patogenezę choroby obejmującej stawy zaangażowane jest wiele czynników, w tym IL-6 i autoprzeciwciała.23 W mazi stawowej pacjentów chorych na SLE i toczniowe zapalenie stawów obserwuje się zwiększoną ekspresję genów zależnych od interferonów.24,25
- nerki: toczniowe zapalenie nerek jest najprawdopodobniej indukowane przez odkładające się kompleksy immunologiczne i aktywację układu dopełniacza. Wynikiem zainicjowanej kaskady zapalnej jest uszkodzenie komórek śródbłonka, infiltracja komórek układu odpornościowego i wzrost stężenia cytokin oraz chemokin. Uważa się, że hipoksja i indukowane przez zapalenie procesy naprawcze nabierają charakteru przewlekłego i przyczyniają się do uszkodzenia nerek oraz zwłóknienia.3,15,26 Jednak rola IFN typu I w tym procesie również jest istotna i obejmuje gromadzenie się plazmocytoidalnych komórek dendrytycznych w obrębie kłębuszków nerkowych oraz ekspresję genów zależnych od IFN typu I w komórkach kanalików (dane z biopsji nerki pacjentów z toczniowym zapaleniem nerek).27,28,29
- naczynia krwionośne: przedwczesna miażdżyca naczyń obserwowana w przebiegu SLE wynika z działania wielu czynników. Główną przyczyną miażdżycy jest najprawdopodobniej uszkodzenie śródbłonka naczyń, a proces ten może być nasilony u pacjentów z SLE za pośrednictwem autoprzeciwciał i cytokin.15,26 W patogenezie miażdżycy naczyń dochodzić też może do aktywacji komórek śródbłonka, która skutkuje: uwolnieniem cytokin, rekrutacją monocytów i powstawaniem komórek piankowatych – w procesach tych rola IFN typu I została jednoznacznie potwierdzona.3,15
- ośrodkowy układ nerwowy: dopiero niedawno odkryto rolę układu odpornościowego (cytokin czy autoprzeciwciał skierowanych przeciw strukturom nerwowym) w patogenezie neuropsychiatrycznej manifestacji SLE.30 U chorych tych zaobserwowano podwyższony poziom IFNα w płynie mózgowo-rdzeniowym.31 Sugeruje się, że IFNγ typu I mogą aktywować komórki mikrogleju, które odpowiedzialne są za fagocytozę struktur neuronalnych i związków wydzielanych w obrębie synaps.30,32
Rolę interferonów typu I w patofizjologii tocznia podsumowuje wideo, które jest dostępne tutaj
Sygnatura genowa interferonów typu I u pacjentów z toczniem.
Interferony typu I stanowią 5 klas cytokin (IFNα, β, ε, κ, i ω), z których IFNα podzielone są na dodatkowe 12 podtypów.32,33 Wszystkie interferony typu I wiążą się z receptorem IFNα, który jest heterodimerem składającym się z podjednostek IFNAR1 i IFNAR2. Miejsce wiązania cytokiny znajduje się na podjednostce IFNAR2, z kolei podjednostka IFNAR1 odpowiada za przekazanie sygnału do wnętrza komórki z wykorzystaniem kinaz JAK/STAT.34
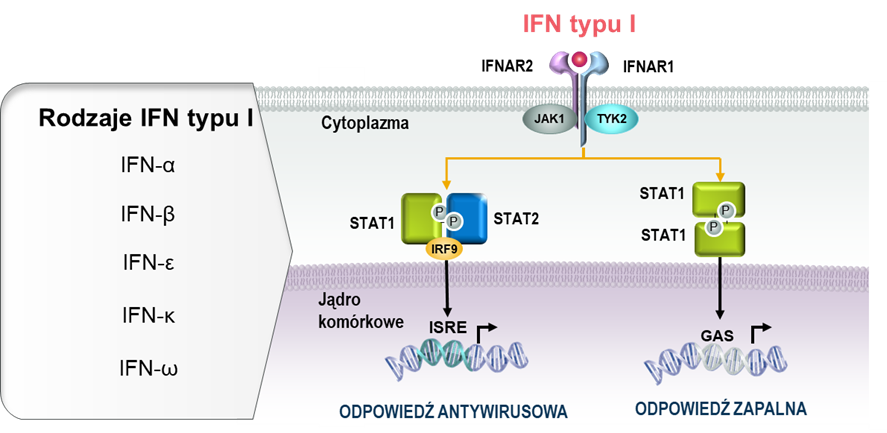
Mechanizm aktywacji ekspresji genów zależnych od interferonów typu I.
GAS = sekwencja aktywowana interferonem gamma; IFN = interferon; IFNAR = receptor dla interferonów α/β; IRF= czynnik transkrypcyjny regulowany interferonem; ISRE = element odpowiedzi stymulowany interferonem; JAK = kinazy janusowe; P = fosforylacja; STAT = czynnik transkrypcyjny; TYK = kinaza tyrozynowa.
Skutkiem wiązania się interferonów typu I do swoistego receptora na powierzchni komórek jest zmiana w ekspresji kilkuset genów, które mają różne funkcje: część z nich odpowiada za indukcję mechanizmów przeciwwirusowych (np. hamowanie replikacji i degradację materiału genetycznego wirusa), natomiast część pełni funkcje immunoregulujące i jest zaangażowana w odpowiedź prozapalną. Zestaw genów, których aktywność jest regulowana przez IFN typu I jest charakterystyczny i można go określić mianem sygnatury genowej interferonów typu I. Sygnatura ta może być różna w zależności od rodzaju badanych próbek; większość badań oparto o analizy krwi obwodowej35, jednak u pacjentów z toczniem zwiększoną ekspresję w obrębie sygnatury genowej IFN typu I zaobserwowano również w skórze, mazi stawowej i nerkach.36 U około 60 do 80% pacjentów z umiarkowaną do ciężkiej postacią tocznia obserwuje się zwiększoną ekspresję genów, które są regulowane przez interferony typu I.15,16,17,18,37
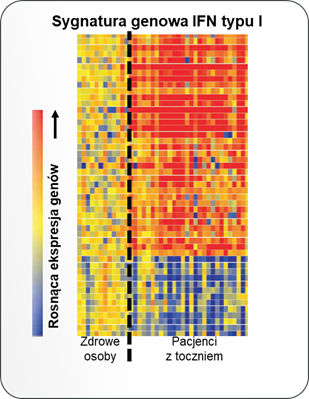
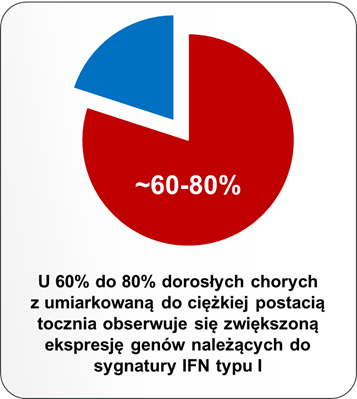
Nadekspresja sygnatury genowej interferonów typu I u pacjentów z toczniem
1. Allen ME, et al. Trends Mol Med. 2021;27(2):152-171.
2. Gupta S, Kaplan MJ. J Clin Invest. 2021;131(3):e144918.
3. Tsokos GC. Nat Immunol. 2020;21(6):605-614
4. Buyon JP, et al. Lupus Sci Med. 2015;2(1):e000087.
5. Lu R, et al. J Autoimmun. 2016;74:182-193
6. Arbuckle MR, et al. N Engl J Med. 2003;349(16):1526-1533
7. Munroe ME, et al. Ann Rheum Dis. 2016;75(11):2014-2021
8. Hooks JJ, et al. N Engl J Med. 1979;301(1):5-8
9. Ytterberg SR, Schnitzer TJ. Arthritis Rheum. 1982;25(4):401-406.
10. Blanco P, et al. 2001;294(5546):1540-1543
11. Bennett L, et al. J Exp Med. 2003;197(6):711-723
12. Baechler EC, et al. Proc Natl Acad Sci U S A. 2003;100(5):2610-2615
13. Crow MK, et al. Autoimmunity. 2003;36(8):481-490
14. Han GM, et al. Genes Immun. 2003;4(3):177-186.
15. Crow MK. J Immunol. 2014;192(12):5459-5468
16. Lauwerys BR, et al. Rheumatology (Oxford). 2014;53(8):1369-1376
17. Hoffman RW, et al. Arthr Rheumatol. 2017;69(3):643-654
18. Becker, AM, et al. PLoS ONE. 2013;8(6):e67003
19. Niewold TB, Swedler WI. Clin Rheumatol. 2005;24(2):178-181.
20. Wenzel J. Nat Rev Rheumatol. 2019;15(9):519-532
21. Sarkar MK, et al. Ann Rheum Dis. 2018;77(11):1653-1664
22. Berthier CC, et al. J Clin Med. 2019;8(8):1244.
23. Ceccarelli F, et al. Semin Arthritis Rheum. 2017;47(1):53-64.
24. Hubbard EL, et al. Sci Rep. 2020;10(1):17361
25. Nzeusseu Toukap A, et al. Arthritis Rheum. 2007;56(5):1579-1588
26. Liu Z, Davidson A. Nat Med. 2012;18(6):871-882.
27. Tucci M, et al. Arthritis Rheum. 2008;58(1):251-262
28. Der E, et al. Nat Immunol. 2019;20(7):915-927.
29. Peterson KS, et al. J Clin Invest. 2004;113(12):1722-1733.
30. Schwartz N, et al. Nat Rev Rheumatol. 2019;15(3):137-152.
31. Shiozawa S, et al. Arthritis Rheum. 1992;35(4):417-422
32. Rönnblom L, Leonard D. Lupus Sci Med. 2019;6(1):e000270
33. Sozzani S, et al. Autoimmunity. 2010;43(3):196-203
34. Ivashkiv LB, Donlin LT. Nat Rev Immunol. 2014;14(1):36-49
35. Barrat FJ, et al. Nat Immunol. 2019;20(12):1574-1583. doi:10.1038/s41590-019-0466-2
36. Catalina, et al. Commun Biol. 2019;2,140
37. Becker AM, et al. PLoS ONE. 2013;8(6):e67003